
{kind=link}
{kind=link}
{kind=link}
青稞籽粒饲草性状遗传特征
[刘新春1  , 赖运平
, 赖运平1, 2 , 余毅3 , 袁金娥1, 2 , 巴桑玉珍1, 4 , 冯宗云1 ]
, 赖运平]
|
|


第一作者:刘新春(1985-),男,四川乐山人,在读博士生,主要从事青稞品质性状分子生物学研究。E-mail:ku0082005@126.com
为了解青稞( Hordeum vulgare var. nudum Hook. f.)的饲草性状的遗传特征并选育具有优质饲草品质和高产的青稞新品种,对60份来源于中国青藏高原的青稞籽粒饲草性状进行研究,利用TASSEL软件的混合线性模型对其进行非条件和条件关联分析,同时结合分离群体群分离法对影响籽粒饲草性状的分子标记进行检测。结果表明,青稞品种间存在丰富的籽粒饲草性状变异;群体结构分析显示该群体可以分成3个亚群,群体的分类结果与品种的来源地有密切的相关性;在非条件关联分析中共发现23个SRAP分子标记和籽粒饲草品质性状(包括粗蛋白含量、淀粉含量、灰分含量)显著关联,而在条件关联分析中20和24 个SRAP分子标记分别和籽粒粗蛋白含量、淀粉含量显著关联。同时利用分离群体群分离法,检测到37个SRAP标记与籽粒的饲草性状相关,其中2个影响籽粒粗蛋白含量,4个影响淀粉含量SRAP标记在非条件关联分析中检测到。该研究结果为青稞的籽粒饲草品质性状的改良和高产优质饲草青稞育种提供依据。
To gain genetic information on forage traits for breeding new hulless barley varieties with high yield and good forage quality, we characterized the grain forage traits of 60 accessions of hulless barleys originating from the Qing-Tibet plateau. Unconditional and conditional association mapping analyses, conducted using a mixed line model as implemented in TASSEL software, and bulk sample analysis (BSA) methodology were used to identify markers significantly associated with grain forage traits. High genetic variation in grain forage traits was detected in the association mapping panel. These barley accessions were classified into three subgroups based on population genetic structure analysis, which indicated significant relationships between accession subgroups and their region of origin. Twenty-three sequence-related amplified polymorphism (SRAP) markers for grain forage traits, including crude protein content, starch content, and ash content, were determined using unconditional association analysis, whereas 20 and 24 SRAP markers significantly associated with crude protein content and starch content, respectively, were detected in the conditional association study. Additionally, a total of 37 SRAP markers associated with variation in grain forage traits were detected using the BSA methodology, among which, 2 and 4 SRAP markers associated with crude protein content and starch content, respectively, were also identified in the unconditional association study. These results provide potentially valuable information for improving grain forage traits and the cultivation of good quality, high-yielding accessions of hulless barley.
青稞(Hordeum vulgare var. nudum Hook. f.)是我国青藏高原藏民族对裸大麦的称呼, 是青藏高原海拔4 500 m以上的地区唯一能正常成熟的粮食作物, 是我国藏族人民的口粮和畜牧的饲料的主要来源[1], 随着近年来藏区畜牧业的发展, 传统的主粮型青稞已不适应藏区畜牧业的发展。因此, 培育具有高产特征、优异主粮和饲草品质的青稞品种对于维护藏区社会稳定和促进该地区经济和社会发展有着重要实践意义。目前有关籽粒饲草品质遗传特征的研究主要集中在大麦(H. vulgare)上, 青稞的相关研究较少, 许多研究者对粗蛋白、酸性洗涤纤维、中性洗涤纤维、酸性洗涤木质素等饲草性状进行了QTL定位研究[2, 3, 4, 5], 但这些QTL都是从连锁作图群体中挖掘出来的, 而且这些QTL通常只代表着少数优质饲草品种的遗传特征, 存在检测时间花费多、育种实践效果差等缺点[6]。
为克服连锁作图方法的缺点, 一种基于群体连锁不平衡的关联分析方法已被大量运用于作物的新基因的发掘与相关优质等位基因变异的鉴定[7]。Cai等[8]利用1 319个DArT标记对158份中国西藏大麦资源材料籽粒总蛋白含量性状进行关联作图, 在6条染色体(除了4H染色体)上检测到了与大麦籽粒总蛋白相关的遗传位点, 这些位点所在区域多数是前人报道的籽粒蛋白QTL的热点区域。Shu等[9]利用大麦的9K iselect SNP芯片对欧洲过去100年的大麦商业品种中的抗性淀粉进行了全基因组关联分析, 在10个与籽粒抗性淀粉相关的SNP的LD衰减区域内, 发现了与籽粒淀粉合成相关的结构基因。随后, 运用相同的方法, 在254份欧洲春性大麦中发现了多个与籽粒淀粉相关性状相关联的SNP标记, 并结合大麦基因组的相关信息, 将这些位点定位于淀粉合成与植物细胞壁合成的相关基因的附近[10]。但这些研究只是对影响单个遗传性状变异的基因进行了遗传解析, 而对这些基因在植物发育过程中和受其他因素影响情况的研究较少, 为更好地解析这些基因的影响类型, Zhu[11]提出的净遗传效应的QTL能够较好地解析这些问题, 并同时提高传统QTL的精确性和完整度。该种方法已在小麦(Triticum aestivum)[12]、玉米(Zea mays)[13]、水稻(Oryza sativa)[14]等多种作物上有大量的运用。最近, Jiang等[15]为了能加快水稻的生育期与产量性状进行分子标记聚合育种, 结合条件QTL和关联分析方法, 发现多个同时影响水稻单株有效穗数和生育期的SSR标记, 而这些SSR标记也在前人的文献报道中出现[16]。而运用条件与非条件关联分析的方法发掘调控青稞籽粒饲草相关性状的QTL(基因)未见相关报道, 因此, 本研究运用该方法解析影响青稞品种籽粒饲草相关性状的遗传基础, 以期为优质饲草青稞品种的选育提供一定的理论基础与实践指导。
供试材料来自中国青藏高原地区60份青稞品种。其中, 25份来自四川, 17份来自西藏, 18份来自青海。其材料编号、名称及来源地参见文献[17], 将试验材料于2013年10月种植在四川崇州国家大麦产业体系基地, 每个品种种植 2 行, 行长 2 m, 株距0.4 m, 行距0.3 m。小区按完全随机区组排布, 共2个重复。肥水管理按正常田间水肥管理进行。试验材料于2014年5月按小区混收。
将每个小区收获的植株混合脱粒, 随机获取200~300粒, 置于博通谷物近红外品质分析仪, 进行青稞籽粒饲草品质相关性状(包括籽粒粗蛋白质含量、总淀粉含量、灰分含量)的非损伤性考察, 每个小区重复3次。
该群体的分子基因型数据为64对显性SRAP标记数据, 具体参见文献[18], 为了降低后续关联分析研究中的假阳性结果出现, 将该群体分子数据中的稀有等位变异频率小于10%的位点剔除。共形成191个SRAP位点。利用Structure 2.3.4软件分析群体结构[19]。软件的参数设定参见文献[20], 并得到相关的Q矩阵。
利用SPSS21.0软件对群体籽粒饲草相关性状表型数据的描述性统计及相关性分析, 用平均值和标准误表示测定结果。采用基于混合线性模型的数量性状分析软件QGAStation[21]对表型数据进行条件化转化。将某个籽粒饲草品质性状数据表现值转化为基于其他饲草品质性状为同一水平的表现值; 以籽粒粗蛋白含量为例, 将籽粒粗蛋白质含量(Crude protein content)分别转化为基于灰分含量(Ash content)、淀粉含量(Starch content)、纤维含量(Fiber content)为同一水平的条件表型值Crude protein|Ash、Crude protein|Starch、Crude protein|Fiber。籽粒淀粉含量以此处理, 并将各个性状的非转化值命名为该性状的非条件化值。
利用博通近红外谷物品质分析仪中的大麦分析模块对青稞品种的籽粒饲草品质性状进行相关扫描分析(表1), 结果表明除了粗蛋白在青海地区呈显著差异 (P< 0.05) 外, 所有的饲草品质性状在各个地区内均呈极显著差异(P< 0.01)。青海地区青稞品种的平均粗蛋白含量最高, 为12.78%。西藏地区品种的平均粗蛋白质含量最低, 为12.20%。总群体的粗蛋白平均含量为12.41%, 而最高和最低粗蛋白含量的品种载体分别出现在地区平均值极差的区域, 即最高含量(18.60%)品种出现在青海的门农1号材料中, 而来源于西藏的藏青21号品种拥有最低的粗蛋白含量(8.00%)。淀粉含量以四川地区的青稞品种最大, 为48.55%, 青海地区次之, 为48.24%, 而西藏地区的材料最小, 为47.44%。总群体的平均淀粉含量为48.14%。而西藏地区材料淀粉含量的极差(4.8)远小于其他两个地区, 来源于青海的北青5号表现出最高的淀粉含量(51.70%), 来源于四川的东升黑材料具有最低的淀粉含量(44.50%)。纤维含量总群体的平均值为6.38%, 西藏地区青稞品种的平均纤维含量最高, 四川、青海地区材料的平均纤维含量低于总体平均纤维含量, 表现为西藏> 四川> 青海, 来源于西藏的藏青21号、QB23表现出最高的纤维含量(7.80%), 而来源于青海的东青2号材料具有最低的纤维含量(5.10%)。对于和纤维具有相似性质的灰分, 来源于四川地区的品种平均灰分含量大于总平均值, 西藏和青海地区品种的平均值较为接近, 且小于总体平均值, 3个地区内的灰分极差均为0.5, 60份青稞品种的灰分在1.45%~2.05%, 以四川地区的俄母1号材料最高, 青海地区的北青8号材料最低。总体来看, 总群体粗蛋白含量的变异系数较大(20.31%), 这说明中国青藏高原青稞品种拥有较丰富的控制蛋白质编码位点, 而淀粉和灰分性状的总体变异系数较小, 可能与这两个性状代表的碳水化合物受到一定的人工选择有关。利用SPSS软件中对籽粒的品质性状进行表型相关性分析(表2), 结果发现, 除粗蛋白含量与纤维含量, 其余的品质性状间均呈现显著(P< 0.05)或者极显著(P< 0.01)相关。
| 表1 青稞品种籽粒饲草品质性状的表型分布 Table 1 Phenotypic distribution of grain forage traits in hulless barley cultivars |
| 表2 青稞品种籽粒饲草品质相关性状的相关性分析 Table 2 Correlation analysis of grain forage traits in hulless barley cultivars |
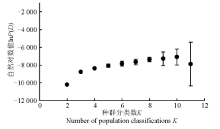
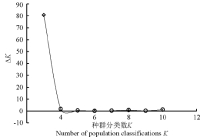
2.2.1 群体结构分析 运用Structure软件对60份中国青藏高原青稞品种的群体分化进行分析(图1), 可以看出, 随着K值的不断增加(2~11), 最大似然值的对数值在不断地增加, 在K=3明显出现一个拐点, 初步认为该群体由3个小亚群构成, 结合Evanno等[26]在2005年提出的Δ K的算法对该群体进行了Δ K发掘(图2)。在K=3时, Δ K峰值出现, 认为该群体应由3个亚群构成。结合其来源地信息, 以Q=0.6为阈值判断该品种所属的遗传类群, 除了阿青4号、阿青5号和拉萨勾芒3个品种属于混合类群外, 其余的57份品种按其来源地, 适当地分布于各自的遗传类群里, 遗传类群与来源地的分布相关性高达100%, 这说明青藏高原的青稞品种的选育与种植地适应性有紧密的联系(图3)。
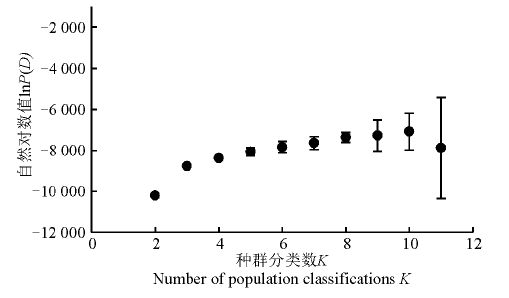 | 图1 K值与lnP(D)值的关系图Fig. 1 Relationship between K and change in lnP(D) values |
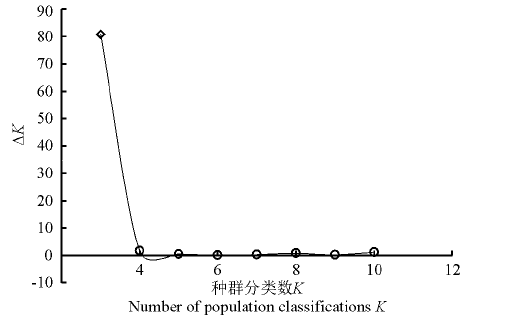 | 图2 Structure软件分析推定的种群分类数K值所对应的Δ K散点图Fig. 2 Δ K for different numbers of populations assumed (K)in the Structure analys |
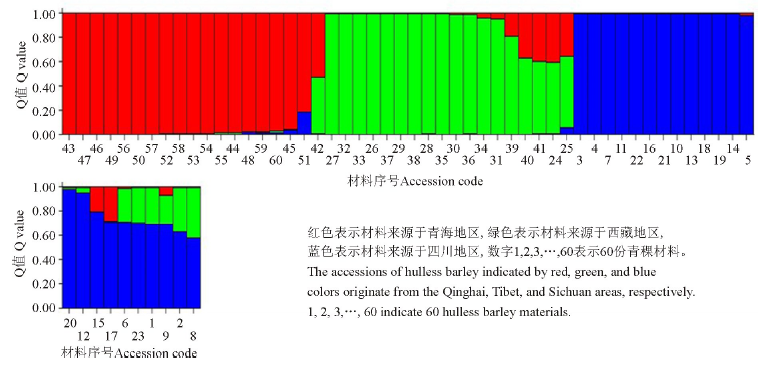 | 图3 利用Structure软件获得的60份青稞品种的群体遗传结构Fig. 3 Population structure of hulless barley cultivars by Structure software |
2.2.2 籽粒饲草品质相关性状的条件与非条件关联分析 运用Tassel软件中MLM模型对青稞品种的饲草品质性状进行非条件关联分析(表3-5)。以P=0.05为阈值, 基于一般混合线性模型共发现了5个控制籽粒粗蛋白含量变异的非条件SRAP标记位点(表3), 5个位点的平均解释表型变异率为9.41%, 其中me11 em14-1位点的R2值最大(13.62%), 其余4个位点的R2相差较小, 在7.34%~8.73%, 以me7 em17-4位点的R2最小。9个影响籽粒总淀粉含量的非条件SRAP标记位点被发掘出来, 9个位点的R2平均值为8.11%(表4), 最高的是me7 em17-4, 为15.13%, 最低的是me10 em15-1, 为5.59%, 除了me7 em17-4位点外, me11 em14-1位点的R2值也大于10%。与淀粉具有同源性的粗灰分含量性状, 也同样发现了9个影响灰分含量的非条件SRAP位点, 9个位点的平均解释表型变异率为7.89%(表5), 范围在5.72%~10.04%, me11 em19-2为最大R2的载体位点, me10 em15-1为最小R2的载体位点。其中me10 em15-1、me6 em13-3和me7 em17-4这3个位点也与籽粒淀粉含量相关联。在P=0.05时, 没有检测到与籽粒纤维含量显著相关的SRAP位点。
| 表3 青稞品种中与籽粒粗蛋白显著相关的SRAP位点及其对表型变异的解释率 Table 3 Sequence-related amplified polymorphism markers significantly associated with grain crude protein and corresponding explained-phenotypic variation in hulless barley cultivars |
运用Zhu[11] 提出的条件QTL的观点, 将相关表型数据进行条件化, 运算得到青稞品种的籽粒饲草品质性状的条件关联标记。对于蛋白含量性状来说(表3), 分别将各种影响籽粒粗蛋白含量饲草品质性状调至同一水平时, 发现了多个与籽粒粗蛋白显著关联的条件SRAP位点, 在将籽粒淀粉含量的影响调整至同一水平时, 分别发现了8个影响籽粒粗蛋白含量的条件性SRAP位点, 这两个性状是所有品质性状中检测到最多的条件性关联位点的性状。纤维则是在粗蛋白条件性关联分析中发掘的最少位点个数(5个)性状。在将淀粉含量调至同一水平时, 发现的8个影响粗蛋白含量的条件性位点, 仅有1个位点在非条件下被检测出来, 而将纤维含量调到同一水平时, 5个条件关联性位点有4个能在非条件下检测出来。对于籽粒淀粉性状(表4), 当消除籽粒灰分含量变异时, 能够检测到最多数目(12个)的影响籽粒淀粉含量的条件关联性位点。而当将籽粒纤维调至同一水平时, 仅有4个条件性位点被检测出来, 2个位点在非条件情况下也能检测, 说明在这2个影响籽粒淀粉的SRAP位点不是通过改变籽粒纤维含量的变异来改变籽粒淀粉含量, 同时也发现在12个去掉籽粒灰分变异而影响籽粒淀粉含量的条件性SRAP位点中, 有8个位点在非条件环境下没有被检测出来, 说明这些调控籽粒淀粉含量是被灰分含量的变异所抑制的, 当灰分变异拉至同一水平, 即灰分因素的影响去掉后, 这些被抑制的位点才能消除抑制影响, 表现出影响籽粒淀粉的表现型出来。
| 表4 青稞品种中与籽粒淀粉显著相关的SRAP位点及其对表型变异的解释率 Table 4 Sequence-related amplified polymorphism markers significantly associated with grain starch and corresponding explained-phenotypic variation in hulless barley cultivars |
| 表5 青稞品种中与灰分性状显著相关的SRAP位点及其对表型变异的解释率 Table 5 Sequence-related amplified polymorphism markers significantly associated with ash content and corresponding explained-phenotypic variation in hulless barley cultivars |
利用QTL-SEQ思路结合群分离基因的观点发掘控制籽粒饲草品质相关的SRAP位点, 以阈值0.3为标准(表6), 分别发现了3个、18个、4个和12个影响籽粒粗蛋白含量、淀粉含量、纤维含量、灰分含量的SRAP位点, me11 em14-1和me11 em17-3两个影响蛋白含量的位点也在非条件环境检测到, me12 em17-1等4个调控籽粒淀粉含量的SRAP位点也在非条件关联分析中检测到, 而影响灰分的SRAP位点均未在非条件关联分析中找到。
| 表6 青稞品种中基于类Δ SNP index 值发掘与籽粒饲草品质相关性状显著相关的SRAP标记 Table 6 Sequence-related amplified polymorphism markers significantly associated with grain forage traits based on Δ SNP index-like value in hulless barley cultivars |
了解青稞品种的遗传组成与遗传背景是改良现有低产品种和选育新的优质青稞品种的前提。随着现代社会的发展, 作物育种家多使用自己熟悉的原始品种和能具有广泛适应性的品种[27], 这客观上造成了当前作物品种的遗传基础狭窄, 以及改良旧有品种的困难。Yang等[18]利用64对SRAP引物对中国青藏高原青稞品种的遗传多样性进行考察, 得到了青稞品种的遗传多样性低, 遗传基础狭窄的结论。这些不同来源地的品种在群体遗传学上呈现什么样的特征, 对于后期的青稞分子标记育种有重要的影响。本研究针对前期的分子数据, 利用基于贝叶斯模型的Structure 软件[19]对青稞品种的遗传构成进行划分, 发现来自于3个地区的青稞品种由3个亚群构成, 除了3个品种外, 其余57个材料分别分成3个亚群。分群的个体与其来源地存在100%相关性。与原来基于遗传距离的聚类分群结果相比, 按群体遗传学分类更能反映出青稞品的遗传基础, 这与魏世平等[28]利用3种分群法探索中国栽培大豆(Glycine max)群体结构时, 得到的基于贝叶斯模型的Structure软件最适宜反映中国栽培大豆的遗传结构的结论一致, 同时也支持了Kline等[29]认为的群体结构分析更能清楚地反映单个个体趋向群体的趋势和各个亚群体之间的基因交流情况。这些结果也反映出中国当代青稞品种的趋势, 即在当地的适应性良好的品种之间杂交选育品种, 而外来资源利用较少, 不同于中国小麦品种在选育过程大量使用少数几个如南大2419等骨干品种[30], 导致品种与来源地存在没有一定的相关性。但同时从Q值矩阵来看(因数据太多, 结果未列出), 发现在归于某些亚群的个体在归类的概率比较接近, 说明在亚群内部的遗传差异较小, 暗示群体内部的遗传多样性较小, 也验证利用SRAP位点发现该群体遗传狭窄的观点。孟亚雄等[1]、赖勇等[31]、巴桑玉珍等[20]分别利用SSR分子标记解析了中国青稞种质资源材料的遗传多样性和其遗传群体结构, 均发现我国青稞种质资源材料的遗传基础差异小, 群体结构与品种的来源地没有联系(即没有清晰来源地亚群的分类), 但是本研究结果(亚群的分类与地理来源密切相关)却与上述观点相反, 可能与本研究利用的材料为品种, 与前面利用的资源材料有所不同。另外, 本研究利用的SRAP标记是一种基于外显子和内含子边界保守性来设计的目的引物[32], 与SSR主要扩增的基因组中的重复区域有所不同, 不同的基因区域一般反映的基因组的信息不同, 功能区域的变异更能准确地反映植物在进化上的适应情况, 因此本研究的分子数据能更良好地反映中国青藏高原青稞品种的差异。
在大量作物QTL定位报道研究中, 发现表型上具有紧密相关性的多个性状的QTL位点多以紧密连锁形式或以一因多效在染色体上分布[33]。Zhao等[34]认为这种一因多效的QTL是一种对于两个关系密切、且又相互独立的性状间的遗传关系的条件QTL的表现形式, Jiang等[15]结合条件QTL定位和关联分析的思路, 发现影响水稻生育期的SSR标记中, 有些是通过影响群体的株高或单株有效穗数的表达来调控水稻生育期的表现, 而有些SSR标记则是直接调控水稻生育期的表现。本研究对青稞籽粒饲草相关性状进行遗传解析, 也发现相似的现象; 即影响籽粒饲草品质性状的条件化关联性状的SRAP位点也大量在非条件化关联分析中找到。因此, 条件性关联分析的结果具有进一步更好地对相关性紧密的多性状的遗传特点进行深层次的解析的能力, 随着高通量测序技术和基因组信息的完善, 这种基于条件化的关联分析结果可以与转录组和基因共表达等数据结合起来, 为动植物复杂数量性状的研究提供一种新的思路。
BSA(bulked sample analysis)是在传统的分离群体群分离法发掘基因的基础上, 结合新一代测序技术发展而来的检测目的性状的功能性分子标记方法, 该方法具有花费少和精确性高等优良特点; 同时运用这种方法的检测标记也不局限于DNA标记, 可以与相关材料的基因表达数据或蛋白表达相结合, 从转录或翻译水平上, 发现与验证与性状相关的标记所联系的功能候选基因[35]。Jin等[36]利用BSA+RNA-SEQ(BSR)方法发掘出了影响茶叶(Camellia sinensis)儿茶素含量变异的重要候选基因F3’ 5’ H基因, 并在儿茶素代谢基因的表达谱数据, 验证了BSR的结果。Zeng等[25]运用BSA的方法, 发现了多个抗柳枝稷(Panicum virgatum)白粉病的SLAF标记, 并在这些标记中发现4个具有NBS结构的R基因。本研究也利用该方法找到69个与青稞籽粒饲草品质相关的SRAP位点, 但与非条件关联分析结果相比, 仅有8个位点是相同的。说明这些位点很可能与青稞品种的饲草品质性状有一定的相关性。但同时也发现了多个未在关联分析筛选出的, 而在BSA方法中获得与籽粒饲草性状相关的SRAP位点, 这可能是由于BSA方法检测相关受到标记种类、混合群体个体数目等多方面影响。针对这些问题, 下一步将对该群体进行多种标记扫描, 并对该群体的饲草品质数据进行多年多点检测, 以期能够找到影响青稞籽粒饲草品质性状稳定表达的遗传位点, 为进一步改良青稞和培育优质饲草青稞品种提供理论依据。
本研究利用遗传群体结构分析了中国青藏高原青稞品种的群体结构, 60份品种分成3个亚群, 群体的类群与品种的来源地密切相关, 非条件关联分析一共发现23个影响籽粒饲草品质性状变异的SRAP标记, 影响淀粉或粗灰分含量的SRAP标记最多, 而在条件关联分析研究中发现44个SRAP标记能影响籽粒饲草品质净遗传变异值。发现6个SRAP标记在BSA分析中和非条件关联分析中均能影响籽粒的饲草品质变异, 这些标记有助于加快青稞籽粒饲草性状的分子育种进程。
The authors have declared that no competing interests exist.
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|